Cardiac Amyloidosis in Autopsy Case of Sudden Unexpected Death
Article information

Trans Abstract
A 78-year-old woman with a medical history of hypertension and diabetes mellitus who underwent surgery for lumbar stenosis died of sudden cardiac arrest two days after the operation. An autopsy was performed; however, the cause of death was not identified macroscopically. Congo red staining detected amyloid deposits in the systemic organs, including the heart, lungs, liver, thyroid, and kidney. Immunohistochemical staining revealed an immunoglobulin lambda light chain, which can cause the primary form of systemic amyloidosis. The prognosis of patients with systemic amyloidosis is directly associated with cardiac involvement. In this case, amyloid formation was noted in the myocardial interstitium and intramyocardial vascular wall, which caused luminal narrowing, subsequently causing arrhythmia and ischemic heart disease in each tissue, respectively. We present a case of primary systemic amyloidosis with severe cardiac involvement that was diagnosed after a comprehensive postmortem examination.
Introduction
Sudden cardiac death (SCD) is most commonly defined as unexpected death from a cardiac cause, either without symptoms or within 1 to 24 hours of symptom onset [1]. SCD is frequently encountered in forensic medicine [2]. The mechanism of SCD is most often a lethal arrhythmia, and coronary artery disease is the leading cause of SCD [1]. However, various cardiac conditions such as cardiomyopathy can also cause SCD. Physiological cardiac conditions such as senile changes can cause SCD in a similar manner [1].
Amyloidosis is a pathological syndrome characterized by extracellular deposition of insoluble protein fibrillary material, defined as amyloid [3]. Amyloid materials are formed by the breakdown of normal or abnormal proteins [4]. There are several types of amyloidosis, and each type causes a different medical condition and prognosis. The storage of amyloid fibrils may occur in various tissues. In cardiac amyloidosis, amyloid storage in the myocardium causes an increase in cardiac wall thickness and mural stiffness, resulting in a gradual onset of restrictive cardiomyopathy [4]. Furthermore, different histological depositions of amyloids can cause varying symptoms [5].
Herein, we present an autopsy case of SCD caused by cardiac amyloidosis. We highlight the typical pathological findings of the cardiac amyloidosis in this case, through the examination of postmortem (PM) cardiac tissue. This case is presented from the forensic pathology perspective.
Case Report
The deceased patient was a 78-year-old woman. She had a medical history of hypertension and diabetes mellitus. She underwent posterior lumbar interbody fusion and posterolateral fusion for lumbar stenosis. She was transferred to a general ward and was found to have cardiac arrest two days after the operation. No issues were identified in her preoperative work-ups, such as chest radiograph, electrocardiogram, transthoracic echocardiogram, pulmonary function test, abdominal ultrasound sonography, and blood laboratory tests (including complete blood cell count, liver function test, and coagulation test).
An autopsy was performed two days after death with a court-issued warrant at the request of the public prosecutor. External examination revealed no injuries other than medical procedures such as surgery and cardiopulmonary resuscitation. On internal examination, cardiac hypertrophy (526 g) with unremarkable ventricular wall thickness (less than 1.5 cm) and mild atherosclerosis of the coronary arteries were noted. However, pulmonary thromboembolism and deep vein thrombosis were not identified, and examination of other internal organs was unremarkable. After gross dissection, the cause and manner of death were assumed to be a SCD of unknown cause and natural conditions, respectively.
Microscopic examination of the heart showed multifocal amyloid deposits in the form of amorphous, eosinophilic, hyaline, extracellular substances. Congo red staining revealed that the amyloids had a pink deposit under light microscopy, and characteristic apple-green birefringence was identified under polarized light microscopy. Immunohistochemical staining of the amyloid protein was positive for lambda light chain but not for kappa light chain, transthyretin, and amyloid A (Fig. 1). Similar to the findings of a previous report [6], amyloid light-chain (AL) deposits were mainly pericellular and reticular. Amyloids may be deposited within any part of the heart, including the endocardium, myocardium, vessels, valves, epicardium, and pericardium. In this case, amyloid deposits were mainly located in the myocardial interstitium and intramyocardial vascular wall, resulting in vascular wall thickening and luminal narrowing. Using Verhoeff-Van Gieson staining, the broken internal elastic lamina was revealed, and amyloid deposits were found to be mainly located in the tunica media (Fig. 2). Furthermore, these amyloid deposits were also noted in the thyroid, lung, liver, and kidney tissues (Fig. 3). However, cerebral involvement was not identified, in accordance with previous findings [7].
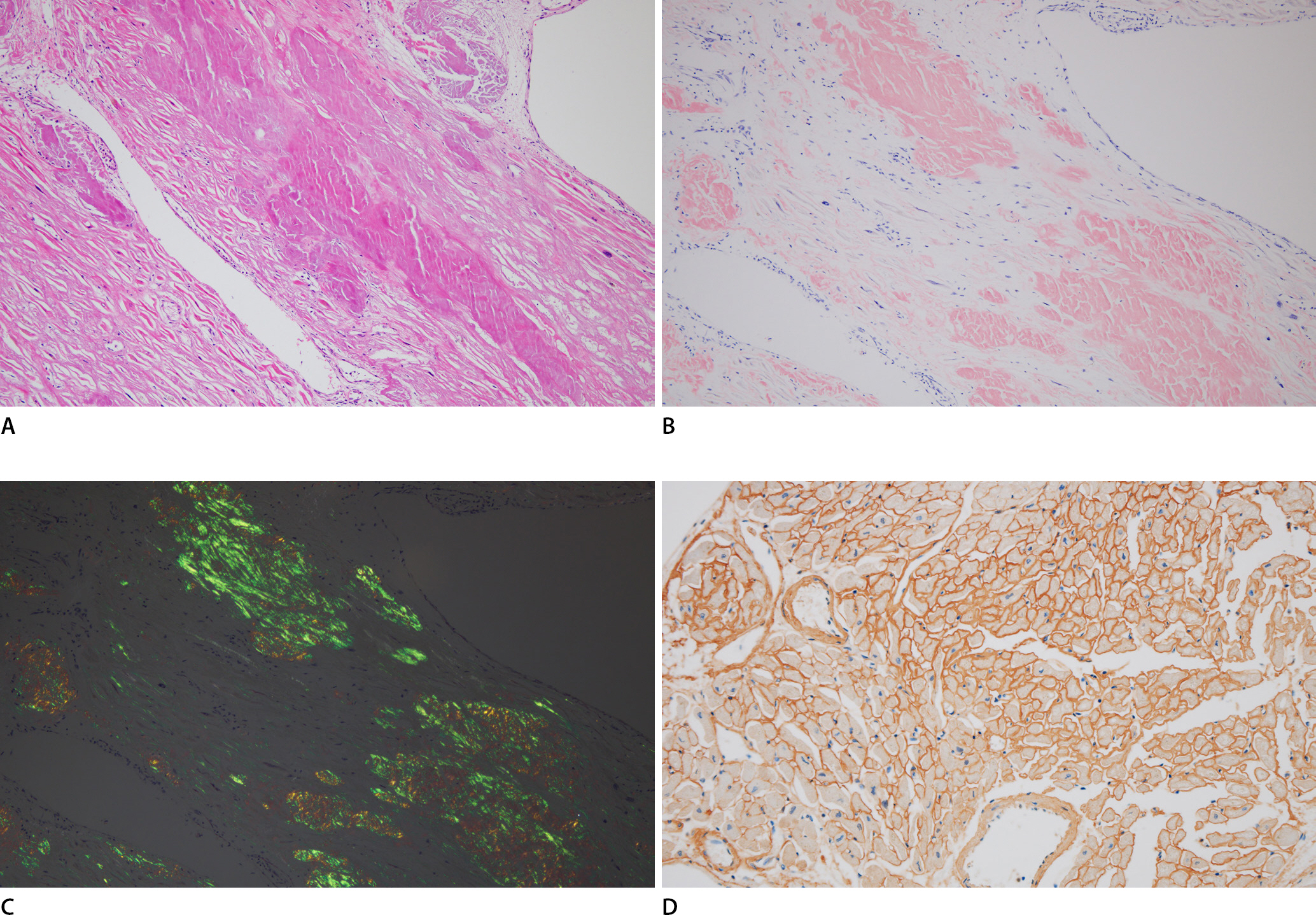
Amyloid deposit is noted in the myocardial interstitium (A, H&E, ×100). Typical amorphous extracellular Congo red stained positive deposits under light microscopy (B, ×100), which display characteristic apple-green birefringence under polarized light (C, ×100), Immunohistochemistry shows immunoglobulin light chain lambda immunoreactivity (D, ×200).
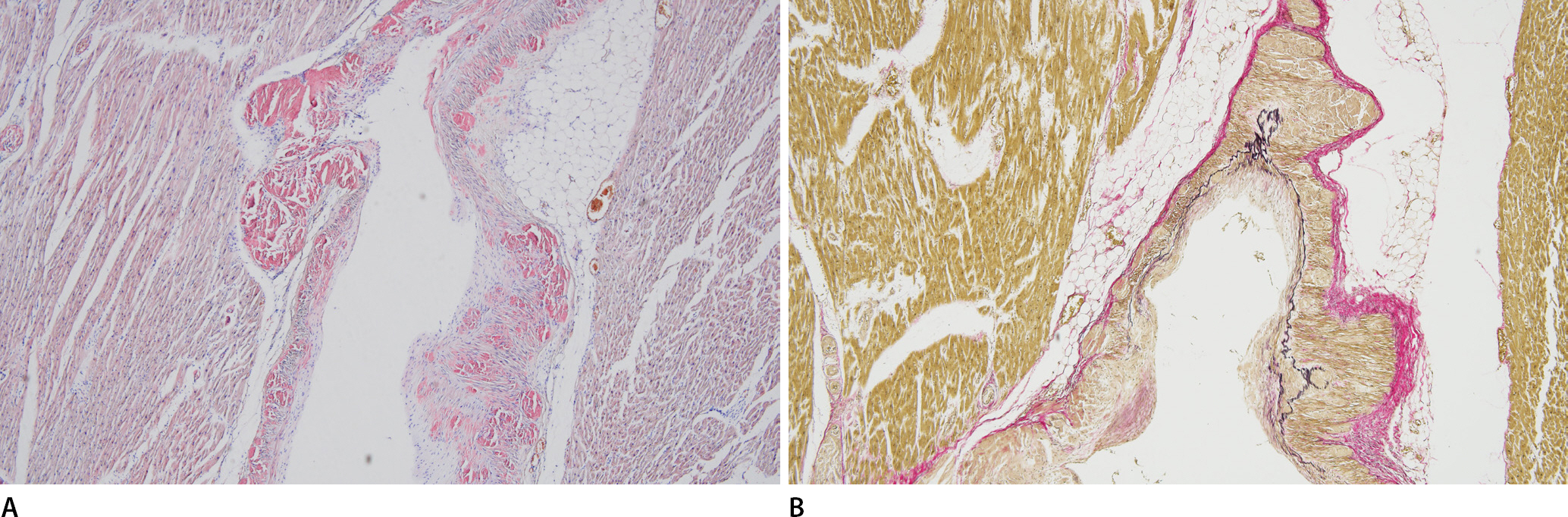
The wall of the intramyocardial coronary artery is thickened by Congo red stained positive material (amyloid) (A, ×100). Verhoeff-Van Gieson elastic staining highlights broken internal elastic lamina and deposition of amyloids in the tunica media (B, ×100).
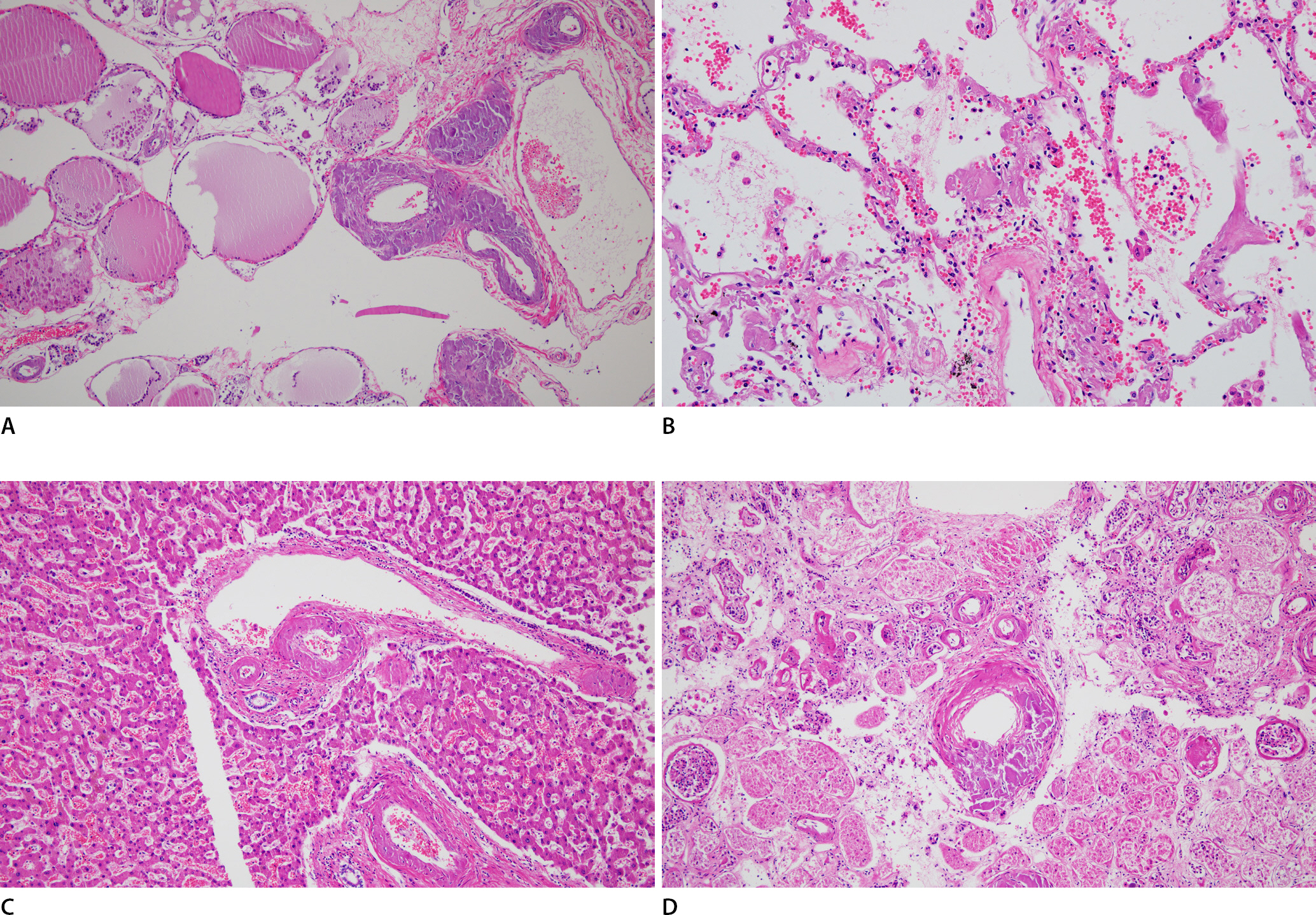
Multiple organ specimens (A, thyroid; B, lung; C, liver; D, kidney), in this case show extracellular eosinophilic, amorphous amyloid deposits (H&E, ×100).
Toxicological tests of PM blood samples were unremarkable. Several inconsequential drugs, such as anti-hypertensive and diabetic drugs, were identified with the blood concentrations within the therapeutic ranges. Blood alcohol concentration in the deceased was less than 0.010%. Biochemical analysis of the vitreous humor and blood ketone levels were unremarkable. The C-reactive protein level was 7.05 mg/dL (reference range, less than 1.0 mg/dL), which was considered to be of postoperative inflammatory status.
After a comprehensive PM examination, including gross dissection, microscopic examination, and PM laboratory tests, we concluded that the deceased died of cardiac amyloidosis in the context of systemic AL amyloidosis. Further, the manner of death was determined to be natural.
Discussion
Amyloidosis is a disease condition associated with several inherited and inflammatory disorders, in which extracellular deposits of fibrillar proteins are responsible for tissue damage and functional compromise. These abnormal fibrils are produced by the aggregation of misfolded proteins that are soluble in their normal folded configuration. The presence of abundant charged sugar groups in these absorbed proteins gives the deposits staining characteristics that are thought to resemble those of starch (amylose). Therefore, the deposits were called amyloids, a name that is firmly entrenched despite the fact that the deposits are unrelated to starch [8]. To date, 37 proteins and peptides that form amyloid deposits in humans have been identified [9].
Amyloid is deposited in the extracellular spaces of various tissues. The symptoms caused by amyloidosis depend on the amount of the deposits and the organs affected. Among these, cardiac amyloidosis can be fatal. Different histological depositions of amyloids can cause different symptoms [5]. In the cardiovascular system, vascular amyloidosis causes vascular fragility, which may lead to bleeding that can occur spontaneously or following relatively mild trauma [8]. Forensic pathologists should consider this condition. Cardiac amyloidosis can occur in various forms of systemic amyloidosis. Amyloids have been identified in several internal organs, including the heart, as in the present case, which was confirmed as cardiac involvement of systemic amyloidosis. The gross feature of amyloidosis in the heart can be unremarkable. Mild enlargement of the heart, along with myocardial wax, pale changes, and rubbery consistency can be observed [3]. Cardiac hypertrophy with an unremarkable ventricular wall thickness was identified in this case. Cardiac symptoms differ according to the deposit location of heart tissue. The deposits begin with a focal endocardial accumulation within the interstitial myocardium.
The electrical conduction system may be affected when amyloid deposits are present in the subendocardial tissue. The most serious consequence of cardiac amyloidosis is conduction disturbances that cause arrhythmias, which can be fatal. Up to half of all patients with cardiac amyloidosis die suddenly, and AL-type cardiac amyloidosis shows conduction abnormalities in approximately 50% of the patients [9]. Cardiac amyloidosis may cause insidious congestive heart failure in the case of interstitial amyloid deposits and produce a restrictive pattern of cardiomyopathy that may masquerade as chronic constrictive pericarditis [4]. If amyloid deposits occur in vascular tissue, they may induce vascular wall thickening and luminal narrowing, which can subsequently cause ischemic heart disease (IHD). Amyloid was noted in the intramyocardial vascular wall, which caused luminal narrowing in this case. The internal elastic lamina was broken and amyloid was deposited in the tunica media. Small intramural coronary vessels were also involved; however, epicardial coronary arteries were spared. Amyloid deposits can affect both the small and large arteries of the heart, and the model describing the formation of amyloid deposits assumes the participation of smooth muscle cells within the tunica media, in which beta-protein precursors accumulate. Furthermore, amyloid deposits were not identified in the small capillaries, suggesting the involvement of the vasculature of smooth muscle cells in the amyloid formation [10]. Many previous reports have described IHD resulting from obstructive intramyocardial coronary amyloidosis [11–15]. We assumed that this patient's cardiac arrest was caused by both a fatal arrhythmia and IHD based on pathologic findings, as well as a history of sudden cardiac arrest during admission after lumbar operations.
The heart is a major organ involved in systemic amyloidosis. In sudden and unexpected cases of death, detailed cardiac evaluation, including microscopic examination, is essential. Forensic pathologists should consider the possibility of cardiac amyloidosis, particularly in elderly patients with SCD. Although all amyloid deposits have similar microscopic findings, “amyloid” is not the same disease as a single chemical entity. More than 30 different proteins can form amyloid deposits, and the three most common forms of amyloid are as follows: (1) AL protein, which is composed of complete immunoglobulin light chains, amino-terminal fragments of light chains, or both; (2) amyloid-associated protein, which is a form of amyloid derived from a unique non-Ig protein produced by the liver; and (3) β-amyloid (Aβ) protein, which constitutes the core of cerebral plaques found in Alzheimer's disease [8]. Octogenarians (and older) also frequently have depositions of extracellular amyloids (most often poorly catabolized transthyretin, amyloid transthyretin [ATTR] amyloidosis) that stiffen the heart and reduce diastolic filling, which is considered a senile change [1]. Cardiac amyloidosis is a rare disease, and it has been reported that this type of cardiac amyloidosis increases with age [14]. AL- and ATTR-type amyloidosis are common in cardiac amyloidosis [4].
The most common systemic amyloidosis is AL amyloidosis, which is caused by plasma cell clones. Amyloid fibrils are formed by antibody light chains in amyloidosis and are precursor proteins [16]. Monoclonal lambda chains are more frequently found than kappa chains in amyloid deposits in primary amyloidosis, whereas monoclonal kappa chains are more frequently found in multiple myeloma or B-cell lymphoproliferative disease without a diagnosis of amyloidosis [17]. AL amyloidosis is a rare condition. Despite its small size, the underlying clone causes rapid progression, often resulting in multiorgan dysfunction through toxic light chains that form amyloid deposits. Clinical manifestations can be clandestine and are often recognized as irreversible [18]. Sudden death is far more common in AL cardiac amyloidosis and can often be the presenting event. AL amyloidosis has the worst prognosis among all the types of systemic amyloidosis. The median survival time is eight months without proper treatment [9]. The clinical symptoms of AL amyloidosis depend on the damage to the affected tissue and organs but are usually non-specific: advanced organ changes most often affect the heart (71%), kidneys (58%), gastrointestinal tract (22%), nervous system (23%), liver (16%), and soft tissues (10%) [19]. Symptoms associated with amyloid angiopathy are diverse and can occur in the form of ischemic disease, myocardial infarction, arrhythmia, malabsorption, proteinuria, and renal failure [20]. Cardiac involvement is a major negative prognostic factor, with 75% of deaths due to heart failure or arrhythmia associated with AL amyloidosis [21]. Further, in the case of AL amyloidosis, nephrotic syndrome is more common, so the kidneys should be microscopically examined, as well as the heart. In addition, when AL amyloidosis is suspected, monoclonal free light chains can be identified in the body fluids, and serum and urine protein electrophoresis and immunoelectrophoresis can be performed [9]. AL amyloidosis was confirmed by the analysis of circulating free light chains, which was performed using diluted PM blood as previously reported [22]. In addition to the concentration of free kappa and lambda light chains, the concentration ratio is important and typically ranges from 0.26 to 1.65 [22].
Finally, we highlight the importance of autopsies to provide a fundamental basis for understanding SCD. After gross dissection, the cause and manner of death were assumed to be SCD of unknown cause and natural death, respectively. Microscopic examination with Congo red staining revealed cardiac amyloidosis with systemic amyloidosis. Furthermore, immunohistochemical staining revealed AL amyloidosis, which could be a cause of systemic amyloidosis. Autopsy literally means self and seeing; in other words, it refers to being an eye witness [23]. PM inspection is undoubtedly the start of PM examination, and autopsy begins with gross dissection. However, this was insufficient. Additional PM tests, such as microscopic examination, including immunohistochemical and special staining, and PM laboratory tests, can reveal macroscopically invisible findings, such as those found in the present case. With a comprehensive approach, the cause and manner of death can be further clarified beyond the visible autopsy findings.
Notes
Conflicts of Interest
Joo-Young Na, a contributing editor of the Korean Journal of Legal Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors declare that there is no conflict of interest.
Acknowledgments
This study was supported by Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital.